1Division of Geological and Planetary Sciences, California Institute of Technology, Pasadena, California 91125-2500, U.S.A.
2Materials and Process Simulation Center (MSC), MC 139-74, California Institute of Technology, Pasadena CA, 91125, USA
ABSTRACT
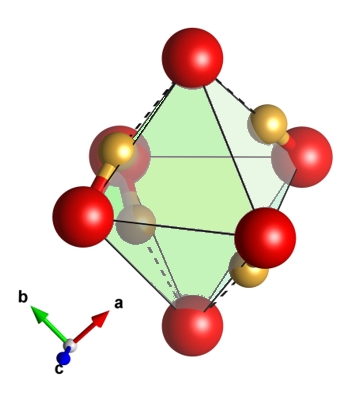
Dense polymorphs of silica have been demonstrated experimentally to incorporate from 1.5% to as high as 11.6% weight percent H2O as OH groups, with important implications for the hydrogen budgets of Earth and other planets. This OH is thought to enter the SiO2 structure via a charge-balanced substitution in which silicon vacancies (VSi) are compensated by protonating four of the surrounding six oxygen atoms, often referred to as a hydrogarnet-type defect. There are many possible configurations for this defect structure in dense silica, but the nature of these configurations and whether they can be distinguished experimentally is unknown. We present here density functional theory (DFT) calculations that systematically assess the possible configurations of a hydrogarnet type defect in stishovite (rutile-type SiO2), with direct comparisons to experimental vibrational spectroscopy data. We predict that stishovite synthesized at 450 K and 10 GPa quenched to room temperature is dominated by a single defect type with tetrahedral geometry. This leads to OH stretching modes (2500-3000 cm-1) and SiOH bending modes (~1400 to 1450 cm-1) largely consistent with the experimental modes. One remaining puzzle is that our calculations produce results largely consistent with experimental data on H to D exchange, but do not explain why a considerable fraction of the 1420 cm-1 mode shifts by only 40 cm-1 in deuterated samples. At elevated pressures and temperatures, we find that a second square planar defect configuration also becomes favorable, leading to modes that should allow experiments to distinguish it from the tetrahedral configuration.