
enstatite (variety hypersthene) from Summit Rock, Oregon
b) Determine the oxidation state(s) of the ion.
There is much evidence to show that Fe2+ is the ion of interest. The stoichiometry of the mineral suggests that divalent cations are the most likely components.
Mössbauer spectra show that Fe2+ is the overwhelmingly dominant oxidation state.
(from Bancroft GM, Burns RG, Howie RA (1967) Determination of the Cation Distribution in theOrthopyroxene Series by the Mossbauer Effect, Nature 213, 1221-1223.)
(Some orthopyroxenes have no Fe3+ detectable yet still show the same spectroscopic features as all other orthopyroxenes.)
c) Determine what sites the ions of interest occupy in the mineral's structure.
We will begin our analysis by considering the sites in which Fe2+ may reside. First we need to look down the c-axis at the orthopyroxene structure. Here is another view looking down the a-axis. Observe what likely sites are available for iron in this structure.d) Determine the point group symmetry of the cation sites.
One way to solve this is to obtain projections of the M(1) and M(2) sites of an orthopyroxene from published X-ray structural data. The projection then enables you to discuss the relative distortions of the two sites in connection with spectroscopic data and site occupancy data. You could use a reference such as Ghose (Zeit. Kristallogr., 122, 81-94, 1965) which refines the structure of an orthopyroxene. Tables 2 and 4 of this paper provide atomic coordinates and atomic linkage data for the pyroxene. Other references are Hawthorne and Ito (Canad. Mineral. 15, 321-338, 1977) and Sueno et al. (Amer. Mineral., 61, 38-53, 1976).
Projections of both an M(1) and M(2) coordination polyhedra on (100), (010), and (001) would be prepared using accurate graph paper or a computer program. The book by Helen D. Megaw (1973) Crystal Structures: A Working Approach" gives directions for preparing the projections. The International Tables for X-Ray Crystallography", Vol. I (1969), provides additional data about the space group and atomic coordinates.
Or you can use a crystallographic program such as the Windows-based program XtalDraw by Bob Downs and explore the structure graphically. You can also obtain from this program's data files or from the American Mineralogist Crystal Structure Database the x,z coordinates of the atoms in the structure.
An easier alternative is to solve this by referring to published structural data. We can use the illustrations in Burns (1970) Mineralogical Applications of Crystal Field Theory, page 90, or page 185 in the 2d edition (1993). In either case, compare the sites and notice their distortions from regular octahedral symmetry.
There is a body of data which leads us to believe that Fe2+ in the M(2) site is responsible for much of the absorption features in the orthopyroxene spectrum. For example, Evans, Ghose and Hafner (J Geology, 75, 306-322, 1967) used Mössbauer spectra and found that most of the Fe is in the M(2) site in low-iron orthopyroxenes. Therefore, we will concentrate on the analysis of the spectroscopy of Fe2+ in the M(2) site.
The point groups determined above, although correct, are not of much use in the detailed analysis of the pyroxene spectra. The problem is that we need symmetry to aid us in the interpretation of all sorts of useful things. Therefore, we need to approximate the symmetry of the M(2) site with a point group of higher symmetry. The reasons for doing this will be obvious only after we work through the entire analysis and find that we can, in fact, do something useful when we are done.
Make the following assumptions: Note that there are three pairs of Fe-O bonds of approximately the same length. Pretend that the two long bonds (2.4 Ĺ and 2.5 Ĺ) are close enough to be the same. Do the same for the two shortest bonds and the two intermediate bonds. Assume further that the four longer bonds are in the same plane and that the two shorter bonds are exactly normal to this plane. You will also have to assume that certain angles are the same; for example, using the terminology of Burns, the angle O4-M2-O3 must be equal to O1-M2-O6.
Chapter 4 in Cotton, Chemical Applications of Group Theory takes you through the concept of the matrix representation of groups and develops the concepts of a character table. You will need to understand these concepts to proceed.
We will be concerned with the character table for C2v in our analysis of the M(2) site of orthopyroxenes.
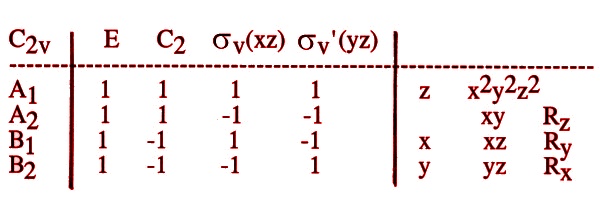
|
Remember, the entries in the character table are the characters of the irreducible representation of the group, the trace of a matrix.
Let’s get some practice using the character table before we get into our ultimate objective.
Problem: Classify the d-orbitals (dz2, dx2-y2, dxy, dxz, and dyz) under C2v.
I’ll do one for you, the dyz orbital:
Remember, the d-orbitals have positive and negative lobes. We will follow the changes in these lobes when we apply the relevant symmetry operations to them. In C2v, there are only 4 symmetry operations left. They are indicated in the character table above.

|
Under the E operation, it returns to +1 times itself.
Under the C2 operation, it returns to -1 times itself.
Under the sv operation, it returns
to -1 times itself.
Under the sv' operation, it returns
to +1 times itself.
By comparison to the character table, we find that anything with these
properties has the symmetry classification B2 in C2v.
Because it is an orbital, we will follow the standard convention and designate
it in lower case letters. Thus, the dyz orbital is a b2
orbital in a C2v environment. Please note that these orbitals are
not oriented in the same way as the d-orbitals of the octahedron; I
did a coordinate transformation so that the orbital z-axis corresponds to
the 2-fold rotation axis of C2v. This is an important point, so
be sure that you understand what happened here.
{kind=link}